Introduction To Pharmacology
#2 of 2
September 1999
Go To Section #1 of 2
Last updated 04/10/00 12:26 PM
Keywords & Concepts:
Elimination
Renal: Most drugs are lipid soluble and diffuse out
of the kidney's tubular lumen when the drug concentration in the filtrate
becomes greater than that in the perivascular space. In order to minimize
this reabsorption, drugs are modified by the body to be more polar using two types
of reactions: Phase I--Involve either the addition of hydroxyl groups or
the removal of blocking groups from hydroxyl, carboxyl or amino groups, or Phase
II--Use conjugation with sulfate, glycine, or glucoronic acid to increase drug
polarity. The conjugates are ionized, and the charged molecules cannot
back-diffuse out of the kidney lumen.
Filtration: Glomerular
filtration –125ml/min. It can’t filter large proteins. Glomerular
filtration happens in the kidneys. Drugs enter the kidney through renal
arteries, which divide to form a glomerular capillary plexus. Free drug
(not bound to albumin) flows through the capillary slits into Bowman's space as
part of the glomerular filtrate. The filtration rate (GFR = 125 ml/min) is
normally about 20% of the renal plasma flow. Lipid solubility and pH do
not influence the passage of drugs into the glomerular filtrate.
Secretion: Drug that
was not transferred into the glomerular filtrate leaves the glomeruli through
efferent arterioles, which divide to form a capillary plexus surrounding the
nephric lumen in the proximal tubule. Secretion primarily occurs in the
tubules by 2 energy-requiring active transport systems, one for anions (weak
acids) and one for cations (weak bases). These systems can transport many
compounds so there may be competition between drugs for carriers.
Note: Premature infants and neonates have incompletely developed tubular
secretory mechanism and thus may retain certain drugs. (Tubular secretion
removes bound and free drug. Proximal tubules carrier systems are active
transport and can saturate. Renal clearance is the sum of filtration and
secretion.)
Reabsorption: As a
drug moves toward the distal convoluted tubule, its concentration increases and
exceeds that of the perivascular space. The drug, if uncharged, may
diffuse out of the nephric lumen back into the systemic circulation.
Manipulating the pH of the urine to increased the ionized form of the drug in
the lumen may be used to minimize the amount of back diffusion and hence
increase the clearance of the undesirable drug. (In the venous blood of
the nephrons – can be active or passive. Things are readily reabsorbed that
have high lipid solubility and a large fraction of uncharged ions at urine pH
and ionized form at plasma pH)
Bile and fecal Enterohepatic
circulation: Brody pg. 43. Can have biliary elimination
then GI tract reabsorption and return to systemic circulation.
Clearance: in general,
what factors affect clearance (e.g., blood flow to organ)
Renal: (filtration,
secretion, reabsorption; These are affected by drug (or metabolite solubility
molecular size, binding affinity to plasma proteins, saturation of transporter
mechanisms, etc…)
Hepatic: Brody pg.
43.
Other:
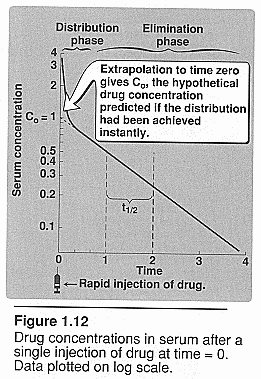
Rate of elimination:
Half life (t1/2): 0.693
Vd/CLtotal Elimination of a drug usually
follows the first order of kinetics, and drops its concentration exponentially
with time. The half life is the amount of time needed to clear 1/2 of the
drugs concentration. (time it takes for [] to decrease to half the values
it had at the start. It is dependent of clearance and Vd)
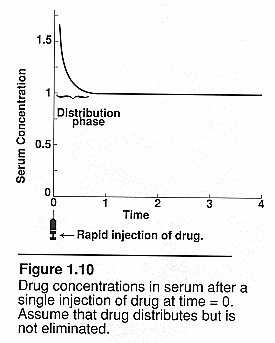
Flow dependent elimination:
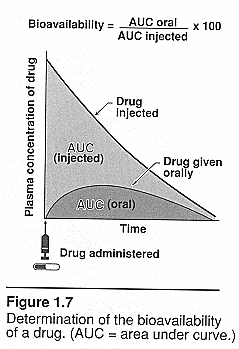
Area under the curve (AUC): Brody pg. 53.
Plasma concentration over time: IV vs oral dosing and clearance. Will depend on
the availability to systemic circulation. IV=CL=dose/AUC for oral=CL=F of
dose/AUC where F = bioavailability See class notes for pictures of curves.
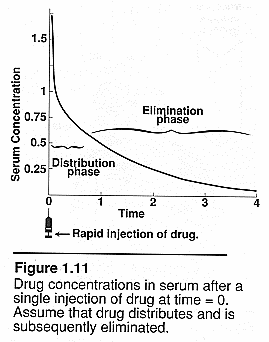
Plasma concentration over
time:
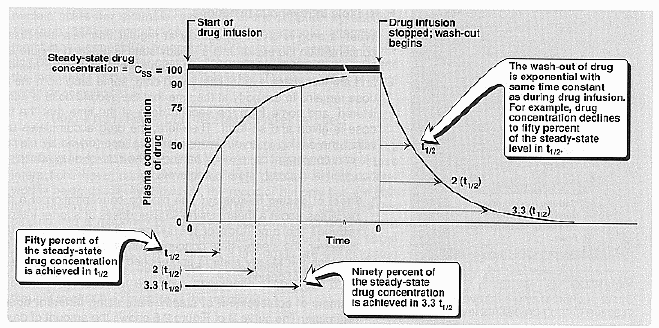
Volume of distribution (apparent): Vd – is a
hypothetical volume of fluid into which a drug is disseminated. Can compare
distribution of drug with volume of water compartment in body. Plasma – drugs
with large molecular wts bind to plasma proteins and are too large to move out
– approx 6% of body wt. ECF – drugs with low molecular wts but are
hydrophilic can move through slits but can’t go through cell membranes. This
is the sum of plasma water and interstitial fluid and is approx 20% of body wt.
Total body water – low molecular wt and are hydrophobic will move freely –
approx 60%
Drug dose and clinical response curves: See
class notes and this page for pictures of curves
Graded dose-response relationships: Brody pg.
31.
Potency: also termed
effective dose [], is a measure of how much drug is required to elicit a given
response. The lower the dose required for a given response, the more potent the
drug. The affinity of the receptor for a drug is an important factor in
determining the potency.
Efficacy: This is the
maximal response produced by a rug. It depends on the number of drug-receptor
complexes formed and the efficiency with which the activated receptor produces a
cellular action.
Quantal dose-effect curves:
ED50: effective
dose at which 50% of the patients responded positively
TD50: Therapeutic
dose where 50% of subjects responded therapeutically to a dose
LD50: Lethal
dose in animal studies where 50% of the animals died at that dose.
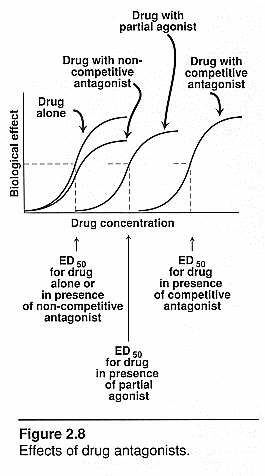
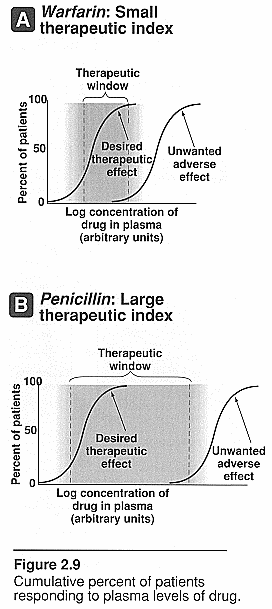
Therapeutic index (TI): this
is the margin of safety of a drug. It is the range of [drug] needed to produce
therapeutic response and one that produces toxic response. The closer this range
the less safe the drug is.
Volume of distribution and clearance (revisited):
Steady-state drug concentration: Brody pg.
58. This occurs when the rate of drug elimination is equal to the rate of
administration. Steady state [] is inversely proportional to the clearance of
the drug.
Rate of infusion: The sole determinant of the
rate that a drug approaches steady state is the half life or drug elimination
and this rate is influenced by the factors that affect half life.
Dosing intervals: The plasma concentration of
a drug oscillates about a mean. Using smaller doses at shorter intervals reduces
the amplitude of the swings of drug [ ].
Half life (t1/2):
Loading dose: Brody pg. 60.
Maintenance dose: Brody pg. 60.
Drug-drug interactions: Brody pg. 77. A
change in magnitude or duration of pharmacological response on one drug because
of the presence of another drug.
Displacement from plasma protein binding
affinity: The example of warfarin is given. This drug is highly
bound to plasma proteins with a high binding affinity. The introduction of
another drug such as an NSAID, which also has a very high binding affinity to
albumin, will now compete with the warfarin for binding sites on the albumin.
This in turn will increase the warfarin free [] in the plasma and will cause
excessive protimes and INR’s since it is free not bound.
Changes in pharmacokinetics:
Age-dependent changes in pharmacokinetics:
Rates of metabolism and elimination: Brody pg.
81. The very old and the very young have either impaired or underdeveloped
methods of metabolism and elimination and dosing needs to be reduced
accordingly.
Volume of distribution: The following factors
affect apparent volume of distribution. How might your estimation of
appropriate drug dose be affected by obesity, excessive tissue edema, or changes
in muscle mass (e.g., elderly)?
Total body water content:
Fatty tissue content and
distribution:
Allergic reactions: Brody pg. 80. Is a
hypersensitization or an undesirable immunological response with drugs acting as
antigens.
Immune responses: Type I –Immediate or
anaphylactoid reaction: IgE antibodies produced by drugs binding to surface of
mast cells and basophils. They cause a release of histamine, leukotriennes,
serotonin, and prostaglandins which trigger rapid immune reactions and cause
bronchial constriction, cap dilatation, urticaria and anaphyl. Shock if severe.
Type II – cytotoxic or autoimmune response – IgG and IgM antibodies and
complement bind to proteins on vascular cell, RBC or WBC or plt surface results
in cytolysis and cell death. Type III – immune complex-mediated reaction –
antigen-antibody complexes interact with and are deposited in tissues like
vascular endothelium or cell membranes promoting acute inflam reaction, serum
sickness, arteritis, urticaria, granulocytopenia. Type IV – cell-mediated
response – delayed. T-lymphocytes, macrophages and neutrophils occurring on
skin combines with skin proteins and cause local inflammation. Halothane induced
hepatitis is an example.
Hypersensitivity: Reaction by a patient who is
more sensitive to a drug (mostly relating to its side effects) then the general
patient population.
Idiosyncratic reactivity: Reaction by a
patient who exhibits an unexpected or unusual response. Most often they are
caused by genetic differences in drug metabolism or an immune response
Developed tolerance: After repeated dosed of a
drug there may develop a state in which the magnitude of response is decreased
by subsequent drug doses of the same size as the initial dose. This
dampened response is called tolerance. Some types of tolerance may be the
result of changes in the concentration of drug at the receptor site and evolve
from pharmacokinetic considerations. Brody pg 32
Tachyphylaxis: Tolerance manifest by rapid
repeated drug administration. (Loss of response in an organ after
repeated exposure to an agonist.)
Go To Section #1 of 2