DAD’s
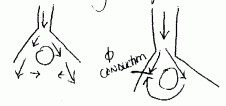
The impulse travels in retrograde direction and re-enters the conduction pathway causing an extra beat. This is the most common cause of arrhythmias.
Understand the basic mechanisms associated with abnormal automaticity and abnormal conduction.
Know the therapeutic uses for each class of antiarrhythmic drugs.
Why do most antiarrhythmic drugs have the potential to be proarrhythmic?
They function to alter the AP or Vmax thru the phases of the
cardiac cycle and depending on whether the channels are open or closed or
inactivated and which drugs are used can have reverse effects. IE. If you block
the K+ efflux the membrane potential stays higher longer and you increase the
incidence of DAD’s and EAD’s. While the cell is depol, the Ca+2
channels are staying open and don’t inactivate quickly which can lead to
triggered activity. Another example is quinidine which activates a
receptors and is vagolytic so you could ![]() VSM
contraction, and
VSM
contraction, and ![]() TPR and potentially
TPR and potentially
![]() HR and if vagal tone is decreased then that could
HR and if vagal tone is decreased then that could
![]() HR and potentiate the arrhythmia.
HR and potentiate the arrhythmia.
Understand the mechanism by which digitalis glycosides and antiarrhythmic drugs can potentiate each other’s toxicity.
Digoxin inhibits the Na/K/ATPase pump which ![]() intracellular Na+ and Ca+2 exchange which leads to
intracellular Na+ and Ca+2 exchange which leads to ![]() intracellular Ca+2 which leads to an
intracellular Ca+2 which leads to an ![]() in
contractility. Digoxin also causes suppression of the AV node, an
in
contractility. Digoxin also causes suppression of the AV node, an ![]() refractory period causes enhanced vagal tone and a ¯
ventricular rate.
refractory period causes enhanced vagal tone and a ¯
ventricular rate.
Lidocaine blocks Na+ ions thus inhibits depolarization and blocks conduction, (¯ action potential specific for vent muscle cells with digoxin and lidocaine together you will ¯ conduction with digoxin at the AV node and ¯ conduction with lidocaine in the ventricular fibers by decreasing conduction in general.
How would the use of certain diuretics (loop and thiazides) potentiate the toxicity of antiarrhythmic drugs?
Loop diuretics inhibit reabsorption in the ascending loop of Henle of Na+/K+/Cl-,
which causes water, Na, Cl, K, and Ca+2 to be excreted.
Antiarrhythmic drugs function as Na+ channel blockers or Ca+2
blockers, or K+ blockers. The massive excretion by a potent diuretic
and the blocking of channels can cause electrolyte imbalances and ![]() arrhythmias and frequency dependent block. where the drug gets in when the
channel is open.
arrhythmias and frequency dependent block. where the drug gets in when the
channel is open.
Last updated 04/10/00 12:26 PM
Return To The MNA 2001 Homepage